发病时间:不清楚
容易跟小儿黏多糖贮积病Ⅳ型混淆的疾病有哪些?小儿黏多糖贮积病Ⅳ型混淆的原因是什么?
补充说明:容易跟小儿黏多糖贮积病Ⅳ型混淆的疾病有哪些?小儿黏多糖贮积病Ⅳ型混淆的原因是什么?
a******7 2011-05-23 09:36
小儿黏多糖贮积病Ⅳ型 黏多糖贮积症Ⅰ型 黏多糖病 生长缓慢 表情淡漠
我要咨询
相关问题
医生回答(1)

于心 主治医师 三甲
提问
容易跟小儿黏多糖贮积病Ⅳ型混淆的疾病如下:
1.黏多糖贮积症Ⅰ型
黏多糖病Ⅰ型有2个亚型,均为α-艾杜糖醛酸苷酶(α-Iduronidase)缺乏症,系因该酶的某种等位基因的突变所致。
一般出生时表现正常。6个月~1岁后患儿逐渐出现生长缓慢,表情淡漠,反应迟钝,智力低下,语言幼稚甚至白痴。大头,前额突出呈舟状,眼距增宽,鼻梁塌陷或扁平,鼻孔增大,唇厚并外翻,张口,舌大且常伸于口外,牙齿小且无光泽,齿列稀疏、不齐。角膜混浊常见,严重者可致失明。
2.黏多糖贮积症Ⅱ型
病因是艾杜糖醛酸-2-硫酸酯酶缺乏。临床上有重型(A)和轻型(B)。由于酶缺乏使硫酸皮肤素(DS)和硫酸类肝素降解障碍,在体内储留并由尿中排出,二者的排出量比为1:1。
较为少见。其中A型的病情较重。患者全部为男性,多于2~6岁起病。临床表现与Hurler综合征相似,但出现时间较晚,进展较缓慢。B型患者病情较轻,有的听力和角膜可均正常,亦无骨骼畸形。
3.黏多糖贮积症Ⅲ型
酶的缺乏各亚型不同。ⅢA型为硫酸酰胺酶(旧名称类肝素-N-硫酸酯酶)缺乏,ⅢB为α-N-乙酰葡糖胺酶缺乏,ⅢC为N-乙酰基转移酶缺乏,ⅢD为葡糖胺-6-硫酸酯酶缺乏
极为少见。虽然本型可有4种不同的酶缺乏,但其临床表现非常相似,主要为进行性的智力减退,其中以黏多糖贮积症ⅢA型的临床进展较快。
4.黏多糖贮积症Ⅴ型
现认为该型即为黏多糖贮积症Ⅰ型的Seheie型,与Hrular综合征不同之处表现为无严重的角膜混浊,且混浊为周边性,患者智力正常,身材正常或稍矮,寿命基本正常,但有多毛,关节强直。背柱、头颅X线示仅有轻微改变。
5.黏多糖贮积症Ⅵ型
黏多糖病Ⅵ型又称Maroteaux-Lamy综合征。为N-乙酰半乳糖胺-4-硫酸酯酶缺乏,临床上分重型和轻型。
极为罕见。临床表现与黏多糖贮积症Ⅰ型相似,但患者的智力正常。一般从2~3岁开始出现生长迟缓。颅骨缝闭合较早,可出现脑积水,并引起颅高压症状和痉挛性偏瘫。角膜混浊出现较早,有进行性听力损害,严重者有失明和耳聋。
6.黏多糖贮积症Ⅶ型
黏多糖病Ⅶ型是β-D-葡糖醛酸酶缺乏,为常染色体隐性遗传,该酶基因位于7q21.2-q22区。
极罕见。特殊面容在出生后不久即开始逐渐出现。一般智力正常,角膜混浊及听力损害较常见。多有肝脾肿大,通常不累及心脏,无腹外疝。上肢较短,骨骼发育不良,可有鸡胸、膝外翻等骨骼畸形。
7.黏多糖病Ⅷ型
黏多糖病Ⅷ型1978年开始报道,病因是由于N-乙酰氨基葡糖-6-硫酸酯酶缺乏,
临床表现有黏多糖病Ⅲ型和Ⅳ型的共同特征,有侏儒,智能落后,脏器受累和骨骼畸形,无角膜混浊。
2011-05-23 09:46
举报向医生提问
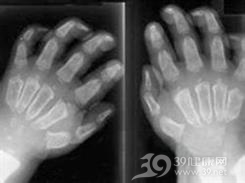
小儿黏多糖贮积病Ⅳ型又称粘多糖增多症Ⅶ型;粘多糖贮积病Ⅶ型;粘多糖病Ⅶ型;戈尔伯杰综合征;β-葡萄糖苷酸酶缺乏症;Goldberg综合征。为常染色体隐性遗传。其临床特点为除头面骨外广泛的骨性病变。20世纪初(Thompson,1900)已有黏多糖病记载,Hurler和Pfaundler(1919)分别报告了精神发育迟滞的儿童中有躯干短小、骨骼畸形的类型。黏多糖贮积病Ⅳ型由乌拉圭Morquio与Breilsford于1929年首先报告。

小儿感冒颗粒
疏风解表,清热解毒。用于小儿风热感冒,症见发热、头胀痛、咳嗽痰黏、咽喉肿痛;流感见上述证候者。
利培酮口服液
1.用于治疗急性和慢性精神分裂症以及其它各种精神病性状态的明显的阳性症状(如幻觉、妄想、思维紊乱、敌视、怀疑)和明显的阴性症状(如反应迟钝、情绪淡漠及社交淡漠、少语)。也可减轻与精神分裂症有关的情感症状(如:抑郁、负罪感、焦虑)。对于急性期治疗有效的患者,在维持期治疗中,本品可继续发挥其临床疗效。 2.可用于治疗双相情感障碍的躁狂发作,其表现为情绪高涨、夸大或易激惹、自我评价过高、睡眠要求减少、语速加快、思维奔逸、注意力分散或判断力低下(包括紊乱或过激行为)。

精苓口服液
补益心肾,养血调肝。用于儿童面色无华,发育迟缓,注意力不集中,记忆力减退,智力低下等症状的改善。

复方苁蓉益智胶囊
益智养肝,活血化浊,健脑增智。用于轻、中度血管性痴呆肝肾亏虚兼痰瘀阻络证。症见智力减退、思维迟钝、神情呆滞、健忘,或喜怒不定、腰膝酸软、头晕耳鸣、失眠多梦等。
健康问答







