发病时间:不清楚
婴儿胆道闭锁
补充说明:婴儿胆道闭锁
l********r 2014-08-27 21:42
我要咨询
精选回答(1)

任正新 主治医师 河西学院附属张掖人民医院 三甲
擅长:擅长糖尿病、心血管系统疾病和临床常见病、多发病的诊断和治疗。对体检综合分析和健康指导有一定的经验。
提问
未结合生化检查数据,但基于典型临床表现如黄疸、陶土色大便和瘙痒,可初步判断为婴儿胆道闭锁的可能性较高。
婴儿胆道闭锁是一种罕见的先天性疾病,其发生与遗传因素有关,也可能是由环境因素影响所致。该疾病会导致胆汁不能进入小肠,引起胆红素在血液中的积累,进而引发一系列症状。
如果新生儿出现不明原因的高胆红素血症,并伴有贫血或脾肿大的现象,则需要考虑是否存在胆管发育异常的情况。
面对婴儿胆道闭锁这一特殊情况,应遵循医嘱进行密切监测和定期随访,同时关注患儿生长发育指标,确保获得适当的营养支持。
2024-03-14 09:06
举报相关问题
医生回答(1)

杨影枫 主治医师 三甲
提问
若怀疑患儿存在胆道闭锁,应当在生后2个月内施行剖腹手术,因为延迟手术会导致患儿发生不可逆的胆汁性肝硬化.术中应作胆道造影以了解胆道情况,肝组织应作活检,冷冻切片了解其形态学改变.仅有5%~10%的患儿能成功地行胆道再吻合术,而其余的患儿通过Kasai术(肝门肠吻合术)常能重建胆汁通道.然而许多患儿术后仍存在明显的慢性病患,包括胆汁淤积,反复胆道炎症和发育迟缓,从而导致晚期死亡率增加.对于肝功能衰竭的患儿,肝移植挽救了肝脏的功能.在此,胆道闭锁是儿科领域最多见的肝移植指征,新生儿肝炎所引起的胆汁淤积通常治疗缓慢,造成永久性的肝脏损伤,一些患儿也因此而死亡。
2014-08-27 21:42
举报向医生提问
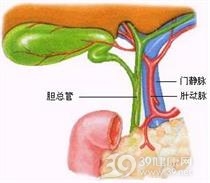
先天性胆道闭锁(congenitalbiliary atresia)是新生儿期一种少见的严重黄疸性疾病,是导致新生儿阻塞性黄疸并需手术治疗的疾病。先天性胆道闭锁并非少见疾病,至少占有新生儿长期阻塞性黄疸的半数病例,其发病率约为1∶8000~1∶14000个存活出生婴儿,但地区和种族有较大差异,以亚洲报道的病例为多,东方民族的发病率高4~5倍,男女之比为1∶2。
健康问答