发病时间:不清楚
考登综合症能不能正常长大
补充说明:考登综合症能不能正常长大
a******W 2022-05-11 18:41
综合症 先天性铜代谢障碍 常染色体隐性遗传病 肝硬化 铜蓝蛋白
我要咨询
精选回答(1)

赵英帅 主治医师 河南省人民医院 三甲
擅长:擅长报告解读、幽门螺旋杆菌感染、高血压、高血脂、高尿酸、冠心病、糖尿病、胃肠炎、便秘、感冒、肺结节、甲状腺结节等疾病的综合诊疗
提问
考登综合症患者不能正常长大。
考登综合症是由先天性铜代谢障碍所致的一种常染色体隐性遗传病,主要表现为特殊的面容、肝硬化、角膜色素环等特征。由于基因突变导致铜蓝蛋白合成减少或结构异常,使铜无法被运输出去,在体内蓄积,造成铜在肝脏、脑、角膜等处沉积,引发一系列临床症状。患者的生长发育受到铜毒性的影响,可能会出现身材矮小的情况。
虽然考登综合症患者不能正常长大,但可以通过治疗来缓解症状,改善生活质量。
建议定期监测患者的铜水平,同时关注其营养状况,确保摄入适量的铜和其他必需微量元素。
2024-02-12 07:33
举报相关问题
向医生提问
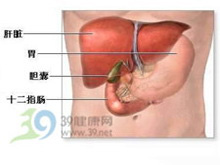
一种是使铜不能合成铜蓝蛋白而广泛沉积在肝、脑、肾、角膜等器官组织中。一种是铜从小肠粘膜细胞运入血液和肝脏中的铜含量减少。
症状起因:1.肝豆状核变性(Wilon病)这是一种常染色体隐性遗传的铜代谢障碍所引起的少见疾病,1912年首先由Wilon报道。本病起因是由于先天性的铜代谢障碍,使铜不能合成铜蓝蛋白而广泛沉积在肝、脑、肾、角膜等器官组织中,引起肝硬化、豆状核变性、肾功能不全、角膜褐色素环,有时可并发急性溶血。血清铜浓度及铜蓝蛋白氧化酶活性降低,尿铜排泄增多。肝豆类核变性者铜吸收增加,达56%。而同时肝脏合成铜蓝蛋白远少于正常,从胆汁排出铜的过程发生障碍,于是引起大量铜在肝脏内沉着,肝铜浓度可达正常人的20~50倍以上。肝内铜沉着超过肝脏铜结合力时,血浆游离铜(非铜蓝蛋白结合铜)就上升至正常的5~10倍,这就导致铜在肝、脑、角膜、肾等器官组织中沉着,造成危害。2.Menke综合征亦称Menke卷发综合征或Menke钢丝样发综合征,是另一种先天性铜代谢异常的疾病。它以X链急性遗传。患者血液、肝脏、大脑中铜减少,组织中铜酶(如细胞色素氧化酶、结缔组织胺氧化酶、多巴胺β-羧化酶和酪氨酸酶)的活力减退,可能是上述病变的基础。
可能疾病:铜中毒引起的溶血性贫血 肝豆状核变性 神经系统遗传病 小儿肝豆状核变性
就诊科室:内分泌

复方吡拉西坦脑蛋白水解物片
1.急慢性脑血管病、脑外伤、各种中毒性脑病等各种原因引起的脑神经功能障碍及记忆力减退。2.先天性脑发育不全,儿童智能发育迟缓,老年性痴呆。3.中枢神经系统感染,病毒性脑膜炎,化脓性脑膜炎,精神病。

利鲁唑片
用于肌萎缩侧索硬化症患者的治疗,可延长存活期和/或推迟气管切开的时间。
前列地尔注射液
治疗慢性动脉闭塞症(血栓闭塞性脉管炎、闭塞性动脉硬化症)引起的四肢溃疡及微小血管循环障碍引起的四肢静息疼痛,改善心脑血管微循环障碍。脏器移植术后抗栓治疗,用以抑制移植血管内的血栓形成。小儿先天性心脏病动脉导管未闭,用以缓解低氧血症,保持导管血流以等待时机手术治疗。

养肝片
对化学性肝损伤有辅助保护作用







