发病时间:不清楚
股骨头软骨病好发几岁?
补充说明:股骨头软骨病好发几岁?
a******W 2019-08-09 14:37
我要咨询
精选回答(1)

陆斌 副主任医师 马鞍山市人民医院 三甲
擅长:熟练掌握四肢骨折的闭合复位以及切开复位内固定术,全膝关节置换术、全髋关节置换术、各类关节翻修手术,半月板成形术、关节清理术。
提问
股骨头骨软化的原因尚不清楚。大多数学者认为慢性损伤是一个重要因素。外伤阻塞骨骺血管,导致继发性缺血性坏死。股骨头骨骺的血供从新生儿到老年变化明显。只有一条外部骨骺动脉供应骨骺。此时血液供应最差。即使是轻微的创伤也会导致血液供应紊乱。年龄增长后,圆韧带血管参与股骨头骨骺的血液供应,发病率开始下降。骨骺板骨化融合后,当骨骺血管进入股骨头时,疾病不再发生。
2019-08-09 15:08
举报相关问题
向医生提问
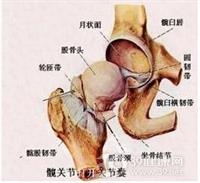
缺血性坏死的特征性病理学改变是由于血液供应受阻而导致的骨细胞死亡。缺血性坏死的严重程度取决于循环系统的受损。股骨头(髋部)是最常见的受损部位;其次为股骨膝关节端和肱骨头(肩部).较少累及踝骨,腕舟骨和足舟骨。股骨头缺血性坏死是由于不同病因破坏了股骨头的血液供应,造成股骨头骨坏死,从而出现髋部疼痛、活动受限等一系列临床表现的一种疾病。可发生于各个年龄段群体,是临床上非常常见的一种疾病。